4 R&D Scientist à Quality Assistance
4.1 Contexte
Je pars au printemps 2015 vers la Belgique, où j’ai occupé un poste de chercheur dans l’industrie pharmaceutique pendant un peu plus de deux ans. J’y ai apporté un profil de chercheur nécessaire pour les projets à mener, accompagné d’une connaissance toute relative de l’industrie pharmaceutique. J’y travaille sous la direction d’Arnaud Delobel, formant une équipe de deux personnes, à laquelle viennent s’ajouter deux scientifiques “prêtés” par le département opérationnel pour six mois chacun: Anicet Catrain et Fabrice Cantais (maintenant Analytical leader for NBE and particularly monoclonal antibody chez Servier) (Figure 4.1). Leur aide précieuse m’aura considérablement facilité la vie, me permettant de me lancer sur mes projets tout en apprenant toutes les particularités de cette industrie très particulière.

Quality Assistance est une CRO, pour contract research organisation, c’est à dire une société à laquelle de grands groupes pharmaceutiques sous-traitent certaines de leurs analyses, le plus souvent en suivant les Bonnes Pratiques de Fabrication (BPF, ou GMP pour Good Manufacturing Practices). Cela implique parfois le transfert de méthodes, du client vers la CRO, ou le développement de nouvelles méthodes. Ces méthodes peuvent être développées pour les besoins spécifiques du client à partir de méthodes génériques, “prédéveloppées” à l’avance et proposées au catalogue de la CRO.
Par exemple, si la CRO souhaite vendre des analyses de peptide mapping, elle peut développer une méthode générique développée sur une ou plusieurs protéines représentatives de ce qui pourraient lui être soumis par un client, et démontrant sa capacité à développer et réaliser ce type d’analyse. Lorsqu’un client soumet une protéine thérapeutique à caractériser, les analystes n’ont “plus qu’à” affiner la méthode. Dans l’exemple du peptide mapping, cela peut être par exemple le choix d’une ou plusieurs protéases adaptées à la séquence (et donc aux sites consensus de clivage) de la protéine du client.
Le contexte régulatoire très strict de l’industrie pharmaceutique est un point de contexte très important à garder en tête dans ce chapitre. Les méthodes doivent être développées dans un environnement BPF très différent des laboratoires académiques, et doivent pouvoir être validées en suivant les guidances des autorités.
J’ai été recruté grâce à un financement FEDER (Fonds européen de développement régional) de l’Union Européenne, pour travailler sur deux grands axes :
Le renouvellement du catalogue de méthodes génériques pour la caractérisation physico-chimique et la quantification de protéines thérapeutiques, et
L’implémentation d’une plateforme d’échange hydrogène-deutérium couplé à la spectrométrie de masse (HDX/MS) pour la caractérisation de structures et interactions de protéines thérapeutiques.
Dernier point divergeant significativement du monde académique : le rythme de développement. Il n’est ici pas question de passer plusieurs mois sur chaque méthode générique. J’ai dédié environ une semaine par méthode, rédaction de la SOP (standard operating procedure) comprise. Il ne s’agit pas ici de passer beaucoup de temps à comprendre pourquoi une méthode fonctionne, mais de la faire fonctionner de façon robuste. De même, lorsqu’une méthode ne fonctionne pas comme souhaité, une approche alternative est rapidement testée.
Pour le second point (HDX/MS), en revanche, j’ai dédié plusieurs semaines à la mise en place de la plateforme analytique puis aux développement de méthodes.
4.2 Développement de méthodes LC/MS pour la caractérisation et quantification de protéines thérapeutiques
Les protéines sont de plus en plus utilisées à des fins thérapeutiques et représentent une part importante des médicaments les plus vendus [1]. Elles comprennent notamment les anticorps monoclonaux et les protéines produites par la technologie de l’ADN recombinant (protéines de fusion, cytokines et facteurs de croissance). Leur caractérisation, du développement au contrôle qualité avant libération des lots, ainsi que la surveillance après la mise sur le marché, est un défi en raison de leur taille et de leur hétérogénéité inhérente au mode de production, notamment en ce qui concerne les modifications post-traductionnelles (PTM) [2–4].
L’analyse d’anticorps et autres protéines recombinantes requiert d’examiner de nombreux paramètres: identités de séquences, nature et position des PTMs, variants de charge, oligomérisation, concentration, structure et interactions. De plus, leur mode de production rend ces paramètres hétérogènes et leur formulation peut être complexe. La caractérisation des variants de ces macromolécules nécessite donc plusieurs méthodes orthogonales, chacune apportant des informations complémentaires aux autres.
J’ai ainsi développé plus de vingt-cinq méthodes pour la caractérisation de protéines et petites molécules thérapeutiques par UPLC couplée à la spectrométrie de masse à haute résolution (ESI-QTOF), aux niveaux intact, des sous-unités, peptidique et des petites molécules (impuretés, leachables). La caractérisation de la glycosylation (Section 4.3) et de la structure et des interactions (Section 4.4) on fait l’objet de travaux plus poussées et sont détaillés ci-après.
Pour ces développements, j’ai principalement utilisé Humira® (adalimumab), un anticorps anti-TNF\(\alpha\) de sous-classe IgG1, approuvé par la FDA et l’EMA pour différentes maladies inflammatoires, dont la polyarthrite rhumatoïde, l’arthrite juvénile idiopathique, le rhumatisme psoriasique, le psoriasis et la maladie de Crohn. L’adalimumab est un anticorps recombinant entièrement humain exprimé dans des cellules CHO. Comme pour toutes les protéines produites par les technologies de l’ADN recombinant, le produit final est un mélange de différents variants. Quand j’entame ces travaux, c’est le médicament le plus vendu dans le monde en 2014, avec des ventes globales de plus de 13 milliards de dollars. J’ai également travaillé avec deux anticorps conjugués (ADC, pour antibody drug conjugate) commerciaux, le brentuximab vedotin (Adcetris®) et le trastuzumab emtansine (Kadcyla®). Ces ADCs diffèrent, entre autres choses, par la nature de la conjugaison aux cystéines ou lysines, respectivement [5].
Pour ces travaux, j’ai couplé de nombreuses phases stationnaires chromatographiques à la spectrométrie de masse haute résolution (ESI-QTOF), qui sert principalement à identifier les espèces éluées. Parmi les phases stationnaires mises en jeu, citons la phase inverse (RP ; confirmation de séquence ; nature, position et abondance des PTMs), l’exclusion stérique (SEC ; détection et quantification de dimères, oligomères et fragments), l’échange d’ions (IEX ; variants de charge), l’interaction hydrophobe (HIC ; quantification des stœchiométries de conjugaison de petites molécules aux ADC), et l’interaction hydrophile (HILIC ; caractérisation de la nature, position et abondance des glycans, voir Section 4.3) [6, 7].
Le couplage direct de certaines de ces méthodes chromatographiques à la spectrométrie de masse n’est pas possible du fait de l’utilisation de tampons et sels non volatils, en particulier la SEC, l’HIC et l’IEX. Une approche classique consiste à collecter des fractions, puis à les dessaler avant l’analyse en MS. Cette méthode est laborieuse car elle nécessite que l’analyste recueille les fractions manuellement ou, s’il utilise un collecteur de fractions, qu’il mesure avec précision le volume de vide entre le détecteur et le collecteur. En outre, la collecte précise des pics peut s’avérer difficile, notamment lorsqu’il s’agit de chromatogrammes mal résolus. Enfin, la génération d’un certain nombre de fractions à partir de chaque échantillon n’est pas souhaitable dans un environnement réglementé, où le suivi des échantillons est essentiel.
Pour remédier à cela, j’ai développé des méthodes d’UPLC bidimensionnelle (2D-LC) en mode heartcutting, mettant en jeu deux vannes, trois pompes et trois colonnes [8]. Chaque hearcut se compose de quatre étapes (Figure 4.2A). L’échantillon est d’abord dirigé vers la première colonne (SEC, IEX, HIC), où les analytes sont séparés, puis détectés par UV ou par toute autre méthode de détection compatible avec la phase mobile (Figure 4.2A-a). Lorsque l’analyte d’intérêt sort de la colonne, une vanne deux positions à six port le dirige vers le flux de la pompe isocratique, puis vers la colonne de piégeage en phase inversée (Figure 4.2A-b). L’utilisation d’une phase mobile aqueuse acidifiée permet de concentrer les analytes sur le piège, tout en éliminant la plupart des sels et en préparant la séparation de deuxième dimension, généralement réalisée avec un système de gradient eau-acétonitrile-acide formique. La vanne rebascule à la fin du heartcut, dont la durée est programmée en fonction de la largeur et de la résolution du pic. Dans une troisième étape, l’analyte d’intérêt est focalisé sur la colonne de piégeage (Figure 4.2A-c). Enfin, une deuxième vanne est activée pour envoyer le contenu de la colonne de piégeage à contre-courant, vers la deuxième colonne analytique, et finalement vers le spectromètre de masse (Figure 4.2A-d).
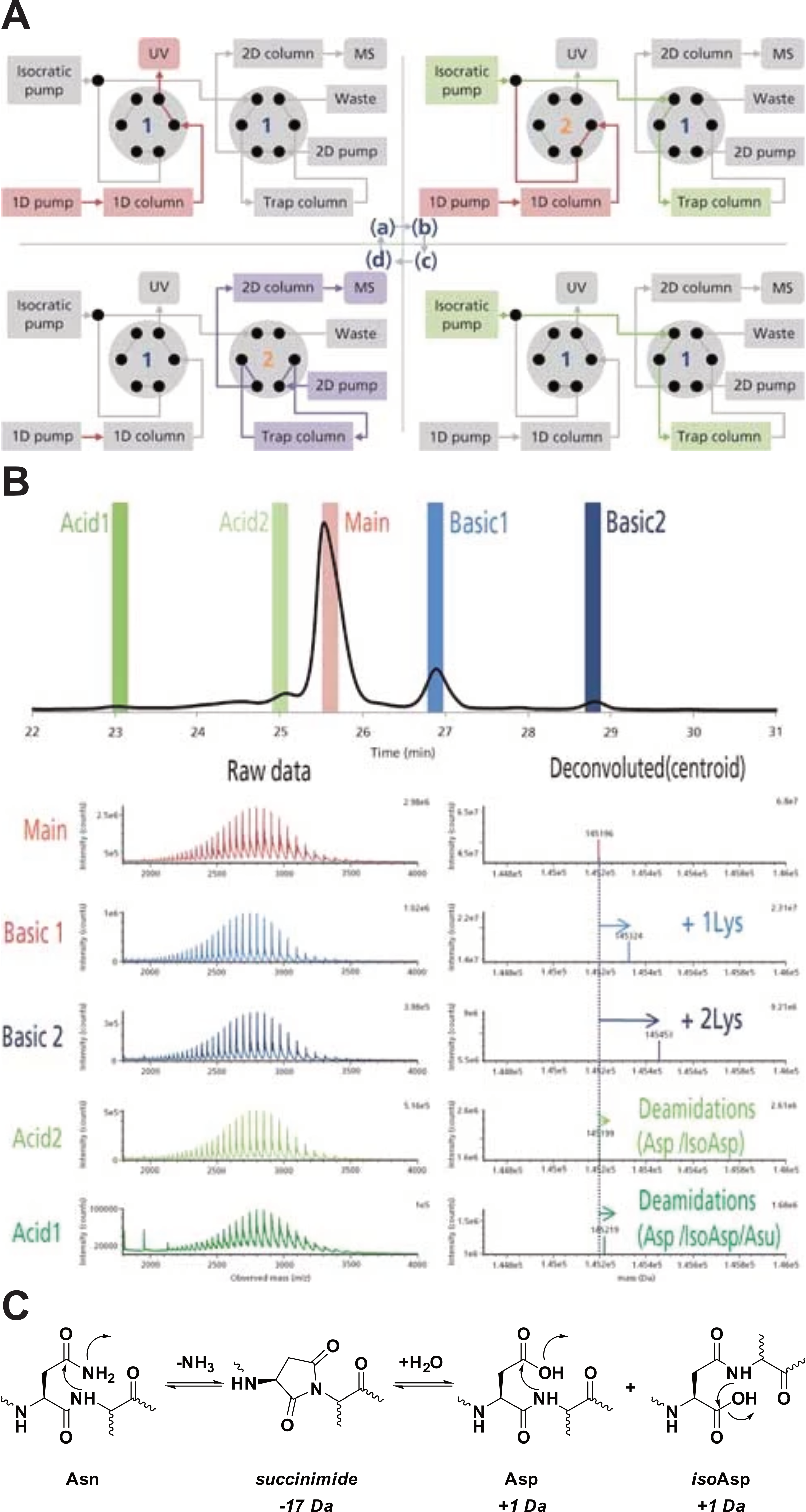
La Figure 4.2B montre l’exemple de l’analyse de variants de charges par couplage IEX-RP/MS, où la combinaisons de ces méthodes apparaît comme particulièrement nécessaire pour identifier des variants de charge. Les variants basiques sont formés suite à la perte de lysines C-terminales, avec une différence de masse assez simple à détecter par MS seule (- 128 Da). En revanche, les variants acides se forment consécutivement à des déamidations de résidus Asn, formant des résidus Asp et isoAsp (Figure 4.2C). La différence de masse entre Asn et Asp/isoAsp est minimale (une unité de masse sur une molécule d’environ 150 kDa !), et la séparation LC est alors très utile. Celle-ci permet aussi de quantifier les différents variants par simple détection UV, étant entendu que les pics ont été initialement identifiés par masse.
La communication de ces résultats dans le milieu industriel m’a ensuite amené à participer à un alpha test de colonnes innovantes pour l’analyse d’anticorps, et au beta test d’un logiciel de contrôle et analyse de données LC-MS, en collaboration avec la société Waters. J’ai également collaboré avec Promega pour le développement de kits de protéases pour peptide mapping et pour l’évaluation de l’activité ADCC (cytotoxicité à médiation cellulaire dépendante des anticorps) [9].
Finalement, ces travaux m’ont amené à utiliser une large gamme de spectromètres de masse (ESI-QTOF, triple quadrupôle, ion-trap ; MALDI-TOF), ce qui m’a permis de renforcer mon expertise sur le sujet, par exemple en mettant au point une méthode LC/MS/MS (MRM) pour la quantification de protéines thérapeutiques diluées dans des fluides biologiques.
4.3 Glycosylation des protéines thérapeutiques
La glycosylation est l’une des modifications post-traductionnelles les plus courantes des protéines et joue un rôle essentiel dans leur fonction, immunogénicité, clairance plasmatique et résistance aux protéases [10]. Par conséquent, le profil de glycosylation des protéines thérapeutiques est considéré comme un attribut de qualité critique, tant pour les innovateurs que pour les biosimilaires, et doit faire l’objet d’une analyse approfondie [11]. Les protéines thérapeutiques sont généralement produites dans différents systèmes d’expression, dont les mécanismes de glycosylation fonctionnent par étapes séquentielles et compétitives, créant ainsi des micro- (nature des glycanes pour un site donné) et macro- (nombre et emplacement des sites) hétérogénéités de glycosylation [4]. En outre, la nature ramifiée des glycans crée un autre niveau de complexité par rapport aux séquences oligonucléotidiques et peptidiques linéaires avec lesquelles j’ai l’habitude de travailler.
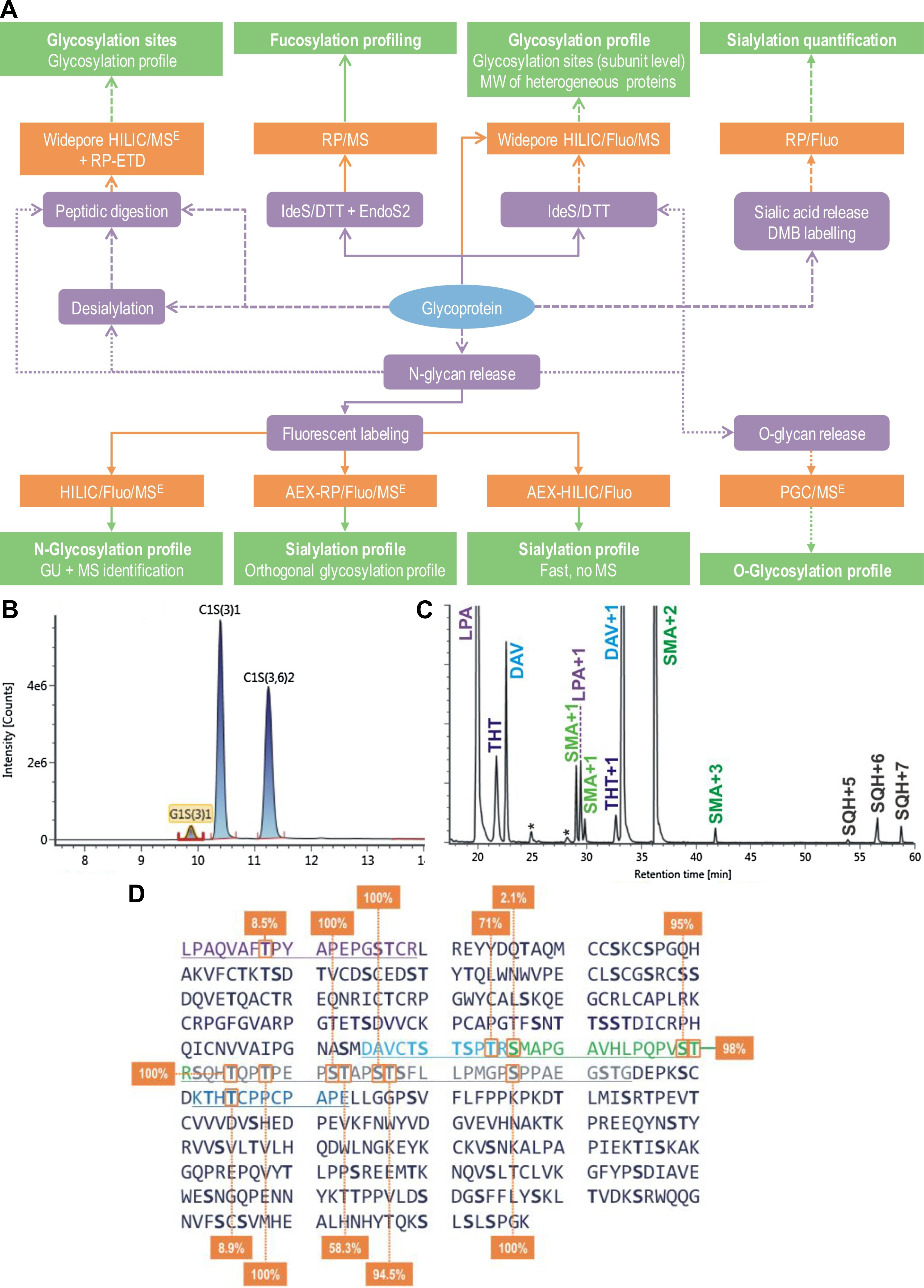
Afin de se conformer aux exigences réglementaires sur cette modification post-traductionnelle de plus en plus étudiée, j’ai proposé l’utilisation d’une combinaison de diverses méthodes LC ou LC/MS(/MS) opérant à différents niveaux d’analyse (glycans libérés, peptides, intactes et sous-unités) simples à mettre en place sans utilisation de matériel et de logiciels hautement spécialisés [11–15]. Ce workflow permet à des scientifiques non spécialisés de caractériser de manière exhaustive les N- et O-glycosylations des protéines thérapeutiques pour, a minima, se conformer aux exigences des autorités. Il comprend des méthodes qui sont ensuite devenues très tendance - en particulier l’HILIC widepore/MS [16–20], mais aussi d’autres peu usitées comme les colonnes à carbone graphite poreux qui présentent une excellent rétention des composés polaires, orthogonale à la phase inverse, et dont nous avons montré l’excellence pour la séparation des O-glycans.
Lors de ce développement, nous avons caractérisé et publié pour la première fois la N- et O-glycosylation complète (natures, sites et occupations des sites) de l’Etarnercept, une protéine de fusion parmi les médicaments les plus vendus au monde [11, 12]. Cela a nécessité l’utilisation de plusieurs techniques chromatographiques, mais également de plusieurs méthodes de fragmentation en phase gazeuse (CID et ETD ; Figure 4.3), et est probablement l’un des objets les plus complexes que j’ai eu à analyser.
4.4 Échange hydrogène-deutérium couplé à la spectrométrie de masse pour la caractérisation de structures et interactions de protéines thérapeutiques
Lors de ma dernière année en poste, mes activités se sont centrées sur l’analyse de structures et interactions de protéines thérapeutiques par échange hydrogène-deutérium couplé à la spectrométrie de masse (HDX/MS). L’HDX repose sur la mesure des cinétiques d’échange des protons amides peptidiques par des deutérons du solvant, qui varient très fortement en fonction de leur structuration [21]. L’échange est ralenti en cas de formation de liaisons hydrogène et d’accès réduit du solvant. L’échange est quantifié par spectrométrie de masse en tirant partie de la différence de masse isotopique entre l’hydrogène et le deutérium.
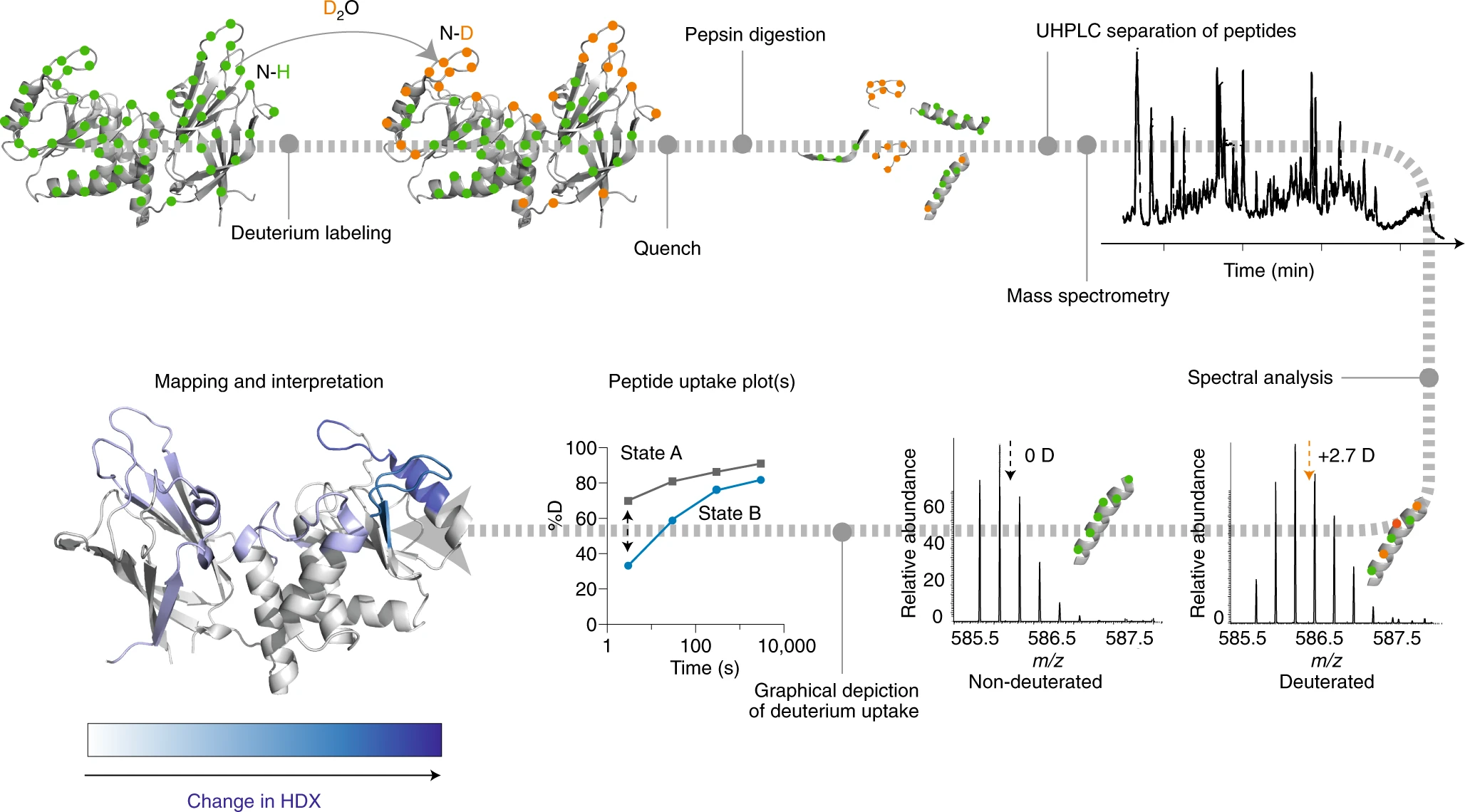
Classiquement, une approche bottom up est utilisée (Figure 4.4). Les protéines sont incubées dans un tampon deutéré pendant un certain temps (d’une dizaine de secondes à plusieurs heures), ce qui permet la deutération des amides. La réaction d’échange est stoppée en abaissant le pH (vers 2.5) et la température. Des dénaturants et agents réducteurs peuvent être ajoutés à cette étape pour dénaturer les protéines. Les protéines sont ensuite digérées par une protéase, généralement la pepsine, à température ambiante. Les peptides sont dessalés et séparés à l’aide d’un système UHPLC réfrigéré, en phase inverse, et dirigés vers un spectromètre de masse, où ils sont ionisés par électrospray et soumis à une analyse de masse. Les niveaux de deutération sont déterminés, généralement en comparant la masse moyenne à la valeur \(m/z\) du centroïde du peptide, pondérée par l’intensité. La cinétique de deutération de chaque peptide peut ensuite être tracée.
Cette approche permet de comparer les cinétiques d’échange de peptides de plusieurs états d’une protéine, par exemple une protéine stressée (haute température, pH non physiologique, oxydant) à cette même protéine non stressée, ou encore un complexe anticorps/antigène à l’anticorps seul. En superposant les résultats de tous les peptides, il est possible d’identifier les sites perturbés (changement structural, complexation non covalente) à l’échelle de l’acide aminé. Les différences de cinétiques HDX peuvent enfin être cartographiées sur une structure de la protéine pour faciliter l’interprétation (Figure 4.4).
Le développement de méthodes HDX/MS n’est pas aisé et dépend assez fortement de l’analyte. En particulier, il est important de bien digérer la protéine, c’est à dire d’avoir une couverture de séquence la plus complète possible, avec une redondance élevée, de façon à ce que les séquences peptidiques partiellement superposée donnent accès à une information résolue à l’échelle de l’acide aminé. Cela est rendu difficile par le fait que la digestion doit se faire très vite (en ligne), à température ambiante et à un pH non optimal, de façon à ce que les deutérons ne soient pas rééchangés par des protons (on parle de back exchange). Cela devient un véritable challenge dans un environnement régulé, notamment de par l’absence de guidances claires sur la caractérisation de structures de protéines thérapeutiques.
Mon premier but a donc été la mise en place la plateforme HDX/MS au sein de la société, une première pour un sous‑traitant analytique européen soumis aux bonnes pratiques de fabrication (Figure 4.1). Dans un second temps, j’ai développé des approches robustes pour la comparaison de lots, les tests de stress [23], et la détermination d’épitopes [24, 25].
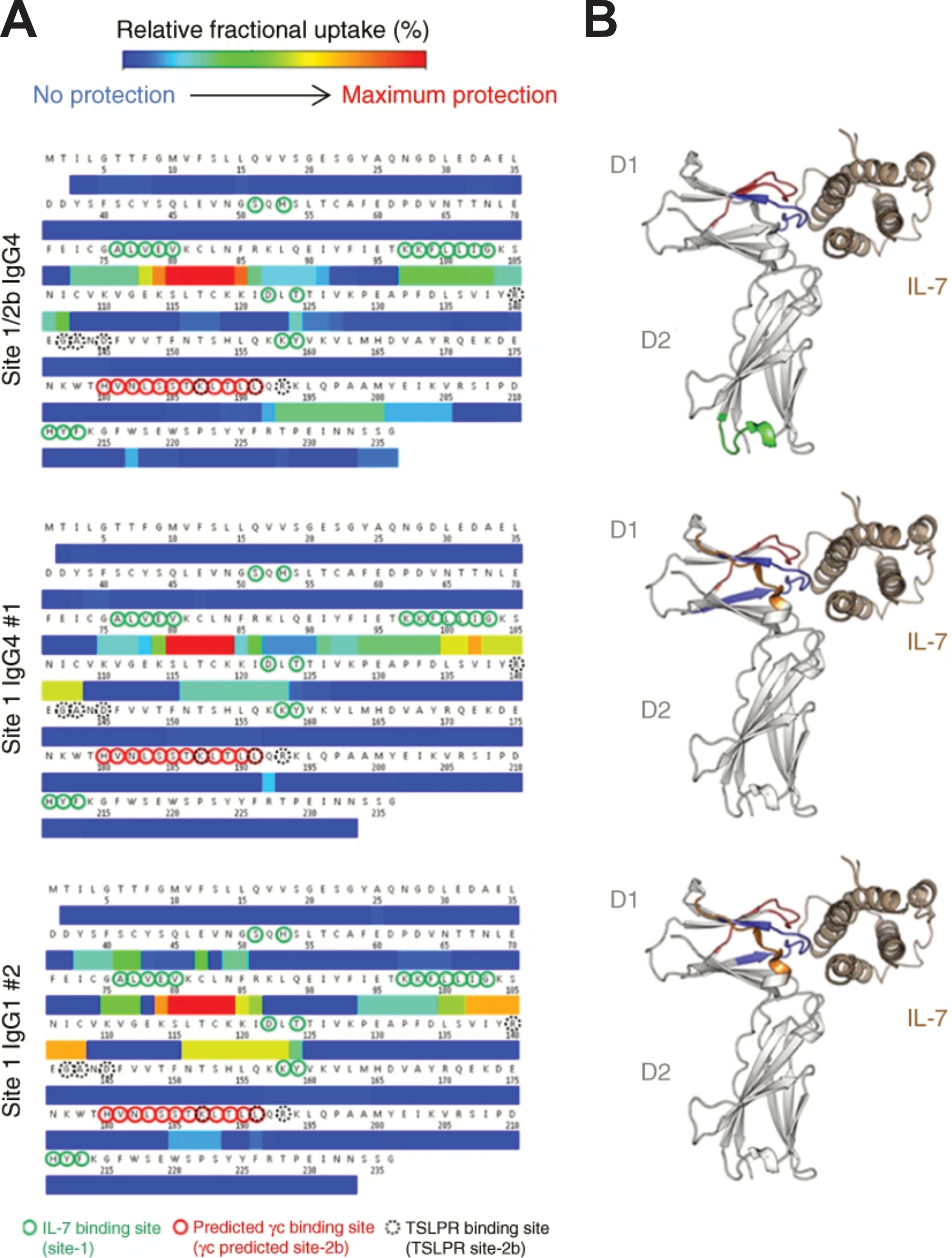
Enfin, j’ai mis en œuvre notre méthode de détermination d’épitope lors d’une collaboration avec l’entreprise Ose Immunotherapeutics (Nantes), qui souhaitait cibler l’expansion des cellules immunitaires « mémoires » pathogènes, comme stratégie thérapeutique prometteuse pour prévenir les attaques auto-immunes chroniques. Dans ce contexte, j’ai déterminé les épitopes de trois nouveaux anticorps monoclonaux anti IL-7R\(\alpha\) par HDX/MS (Figure 4.5) [26]. Grâce à cela, il a été montré que les anticorps liant le domaine d’interaction de l’IL-7 (site-1) ont des propriétés à la fois agonistes et antagonistes, tandis qu’un anticorps liant aussi le domaine d’hétérodimérisation de l’IL-7R\(\alpha\) /\(\gamma\)c (site-2b) présente une activité antagoniste stricte. Une seule injection d’anticorps monoclonaux antagoniste peut induire un contrôle à long terme de l’inflammation cutanée malgré des provocations antigéniques répétées chez des singes présensibilisés.
Ose Immunotherapeutics avait déjà identifié les épitopes par une autre méthode avant de nous contacter, sans toutefois nous en informer. J’ai donc réalisé l’analyse de manière indépendante, à l’aveugle, ce qui a naturellement renforcé notre confiance dans les résultats lorsque Ose nous a confirmé avoir obtenu les mêmes régions.1
1 À mon plus grand soulagement…
4.5 Conclusion
Ces années passées dans l’industrie m’ont été très profitables, puisque j’ai pu travailler sur de nombreuses techniques dans un environnement très différent des laboratoires de recherche publiques, tout en conservant une activité académique.
J’ai ainsi travaillé avec un très large éventail de méthodes de caractérisation et de quantification des protéines et des petites molécules thérapeutiques, ce qui a renforcé mes compétences techniques. Les différences de moyens et de temporalité avec le monde académique ne m’étaient pas inconnues, ayant une formation initiale plutôt industrielle, mais cette nouvelle expérience m’a conforté dans mon choix de carrière académique. J’ai apprécié l’efficacité du travail mise en place, mais j’ai regretté le relatif manque de liberté dans les orientations scientifiques et le peu de discussions techniques poussées. Tout cela est parfaitement compréhensible au regard des objectifs d’une société comme Quality Assistance, bien entendu.
Travailler dans un contexte réglementaire très strict, bien que chronophage et, soyons honnête, parfois pénible, a également été très formateur. Bien que ne travaillant plus dans un environnement GMP, j’essaie de conserver les bonnes habitudes que j’ai acquises à cette occasion. Et je tiens par ailleurs en très haute estime le système qualité de Quality Assistance. Cette expérience constitue également une base importante de mes enseignements, ainsi que de la formation continue que j’offre aux étudiants que je supervise au laboratoire (bonnes pratiques de fabrication, connaissance du monde industriel).
Enfin, bien que les travaux que je résume ci-dessus n’impliquent pas d’acides nucléiques, ils ont été une source d’inspiration pour l’analyse de ces derniers, en particulier l’utilisation de l’HDX/MS pour la caractérisation de structures d’oligonucléotides et de leurs interactions avec des protéines et des petites molécules.